Research in the Rodríguez-Muela Lab:
The driving objective of our research is to discover the cellular and molecular mechanisms underlying the selective neuronal death that characterizes neurodegenerative diseases (NDs). Based on our research, on observations from post-mortem human brains and on recent compelling findings in the field, we propose that this selective death is not stochastic but governed by intrinsic and extrinsic processes. Further, we hypothesize that, in genetic NDs, intrinsic factors play an initiator role while extrinsic factors have a contributor function. Finally, we postulate that neurodevelopmental abnormalities and proteostasis dysregulation are main triggers of the selective pathological cascade in genetic NDs. We address these questions on human models of the motor neuron diseases (MNDs) SMA and familial forms of ALS and familial forms of Alzheimer’s disease (AD).
1. Investigating the source of SMN heterogeneity among MNs, a prediction of MN survival fate.
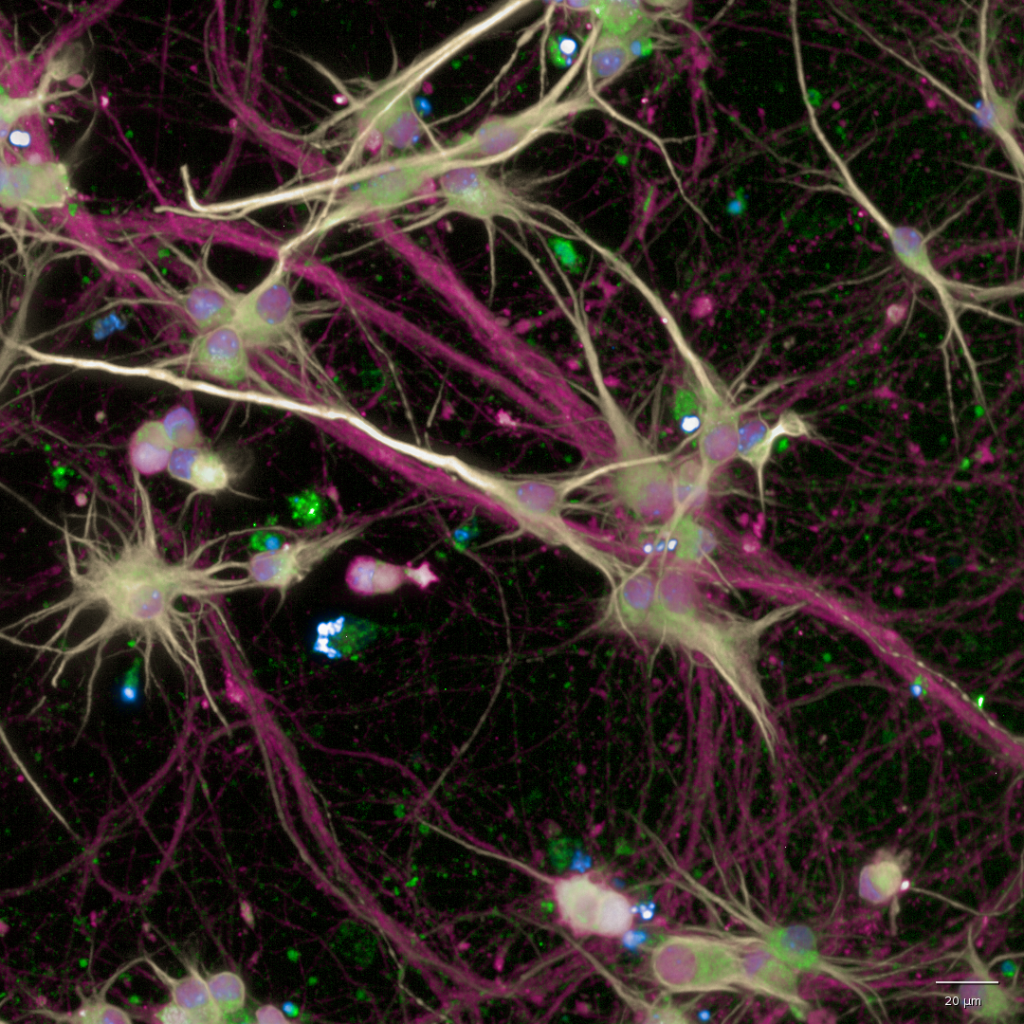
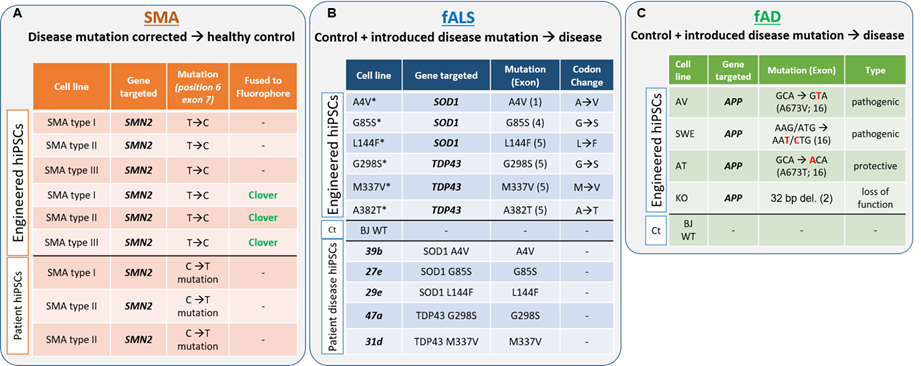
SMN is a highly conserved, ubiquitously expressed protein, located in the cytoplasm and in nuclear foci called gems. When fully absent, cells die. Deletion of SMN in mice is lethal at the morula stage, while hypomorphic mutations in the SMN1 gene lead to SMA of varying severity in humans. Two essential functions for SMN have been described thus far: biogenesis of spliceosomal small nuclear ribonucleoprotein particles (snRNPs), central for mRNA splicing, and axonal transport of mRNAs. However, these functions do not explain why MNs are selectively affected when SMN levels are deficient and why MNs sharing developmental programs and functional properties respond differently to SMN loss. We have found that SMN protein levels are not constant among MNs within the same individual, healthy or diseased. Importantly, in both SMA and fALS, lower levels of cellular SMN are associated with an increased likelihood of MN death. However, this link was only based on correlative findings. Taking advantage of the SMN:Clover hiPSCs lines that our group has recently generated (both healthy control and disease SMA and fALS), and using time-laps longitudinal imaging couple with unbiased automated quantifications, we can now demonstrate that a direct association between SMN protein levels and MN survival exists. These results are of high value since they can help us to predict which is the SMN threshold for survival and which are the MNs that will respond to SMN-increasing treatments in each patient. Now, we are pursuing multi-omics approaches to tack the origin of SMN MN heterogeneity and decipher how are the intrinsic high-expressors different from the low, with the ultimate goal to cenvert SMN-expressors into high before thez become vulnerable and degenerate.
2. Exploring whether a dysfunction of the lysosome-autophagy axis renders low-SMN MNs selectively vulnerable.
The lysosome-autophagy axis is the catabolic pathway in charge of degrading and recycling protein aggregates and unnecessary or damaged organelles through the fusion of autophagosomes (APs) with lysosomes. Its role in maintaining cellular homeostasis is vital in neurons, due to their postmitotic and highly specialized nature, and its dysregulation leading to the accumulation of undegraded material has been implicated in an astonishing number of NDs. Alterations in ubiquitin-dependent proteolysis, mitochondrial function or extracellular matrix among others have been suggested as contributors to selective neuronal loss in MNDs. However, other studies propose that changes in those pathways are not restricted to the affected neuronal type. This controversy is the basis for the theory that therapies need to be tailored for different cell types and stages of the disease. Cytoplasmic aggregates composed of disease-associated proteins have been widely reported in ALS-affected neurons. On the contrary, SMA had been traditionally excluded as a protein-aggregation disease, until recently, when we showed that SMN deficiency leads to an abnormal over-activation of mTOR resulting in a defective AP clearance and accumulation of autophagy substrates. How SMN controls autophagy activity is a totally unexplored field of tremendous relevance, given SMN essential role in promoting MN survival and the required proper functionality of autophagy for neuronal homeostasis. We have recently discovered that only the vulnerable MNs display this lysosomal failure which could render them to degenerate early during the course of the disease. These are novel findings of important clinical impact since, once observed that the current SMA drugs (splicing modifiers or gene therapy) do not represent a cure but a life-long treatment and that there is high variability in the patient response to these drugs, combinatorial therapies are the path forward and lysosomal functionality could be a target. We are now digging deeper into how mechanistically, SMN, mainly known for its role in mRNA splicing, controls the functionality of the lysosome-autophagy axis.
3. An isogenic organoid-based model to study resilience vs vulnerability in early onset Alzheimer’s disease.
Selective neuronal vulnerability has also been extensively described in AD. Image-based studies in patients have revealed that pyramidal neurons in layer II of the entorhinal cortex, in hippocampal CA1 and subiculum are highly vulnerable in early AD, while spinal MNs, neurons in the brain stem, cerebellum, and basal ganglia function properly. Previous studies suggested that damage in a selective population of affected neurons could be the primary driver initiating pathological changes in AD brains. However, the molecular underpinnings of such selective death as well as the order of events that lead to it remain elusive. Furthermore, indications of a neurodevelopmental component are just starting to surface for AD.
Our model recapitulates several AD hallmarks, including abnormally high amounts of Aβ40-42, faster death of cortical neurons (CxNs) than other neuronal types and Aβ extracellular aggregates, an aspect that has been very challenging to achieve to date using hiPSCs or any other cellular model without overexpressing several APP and PSEN1 mutations simultaneously. We are using our isogenic APP-AD model to 1) understand how different Aβ and C-terminal APP fragments resulting from the different APP familial AD-linked mutations have a distinct biological impact in CxNs; 2) uncover the earliest pathological events to find new biomarkers to better detect and understand AD and 3) investigate whether mutations causing early-onset fAD lead to pathological developmental states that might be the underlying trigger for the emergence of the vulnerable phenotype.
4. Investigating whether developmental abnormalities underlie selective vulnerability in NDs.
Accumulating evidence suggest that potentially all NDs have a developmental component that could be essential for neurons to manifest disease hallmarks. The molecular nature of the striking differential vulnerability that characterizes NDs remains a mystery, hindering a full understanding of neurodegeneration and the discovery of efficient therapies. Using spinal cord and cortical organoid models and our cohorts of isogenic SMA, fALS and fAD hiPSCs, we have several fascinating projects focused on studying these diseases from a developmental angle.
One of these studies got recently published in Cell Reports Medicine (https://www.cell.com/cell-reports-medicine/fulltext/S2666-3791(24)00373-2). https://www.cell.com/cell-reports-medicine/fulltext/S2666-3791(24)00373-2). Here, using our new isogenic SCO model, we have discovered fate commitment defects of neuromesodermal progenitors during the early embryonic development in SMA, long before motor neuron loss. Importantly, we have validated these findings in the most commonly used SMA mouse model
(https://idw-online.de/de/news837533,
https://www.genengnews.com/topics/translational-medicine/spinal-muscular-atrophy-organoid-study-points-to-early-neurodevelopmental-defects/
https://www.sciencedaily.com/releases/2024/07/240726113318.htm
https://www.dzne.de/en/news/press-releases/press/prelude-to-neuromuscular-disease-sma-may-offer-chances-for-better-treatment/
https://tu-dresden.de/tu-dresden/newsportal/news/vorgeschichte-der-nervenerkrankung-sma-koennte-chancen-fuer-bessere-behandlung-bieten).
If successful, this line of research has the potential to not only drastically change our understanding of these NDs, but also open completely new therapeutic avenues.